

欧盟是一个对农药有着稳定需求量的地区,据欧盟统计局统计,2011年到2018年,欧盟的农药销量一致保持在每年大约3.6亿公斤的水平。此外,欧盟的成员国中,像法国、西班牙、意大利和德国,不管是从农业发达水平还是农业规模上讲都是处于世界领先地位的。可以说,欧盟的农药领域还是十分具有市场发展潜力的,非常适合进行战略布局和发展。

来源:欧盟统计局
农药企业可以怎么进入欧盟市场呢?性价比最高的方式就是做原药的等同性评估,即TE评估(Technical equivalence assessment)。TE评估周期短,费用低,一般企业只需花费数万元人民币,几个月到一年就可以拿到批准信,产品便可以进入欧盟市场。
TE评估可谓是企业进军欧盟市场的敲门砖,但门打开了,企业想要更好的进行市场布局,仅仅靠敲门砖就有点不够了。根据欧盟农药法规,终端制剂产品的登记需要同时提供原药和制剂两套数据,制剂厂商一般通过购买有数据的原药来源,同时获得全套的原药数据来支持制剂登记。而通过TE评估进入欧盟市场的原药来源是没有全套原药数据的,无法支持制剂客户进行制剂登记,由此,在销售上会有一定的限制。一般只能销售给本身有能力完成登记的制剂客户作为新增原药来源,销售利润也会有所受限。如果想要在欧盟市场占据更大的主动权,则需要参与原药再评审。
参与原药再评审后,再评审通过的原药来源会成为参考来源,不仅能在欧盟合法销售,还因为拥有全套原药数据,销售上会有更大的自主权。在后续的贸易中,可以自由挑选客户,在获取客户的时候会比作为TE来源时相对容易,在销售中也能获得更高的利润。
此外,参与再评审,可以随时详细了解再评审的情况,避免首家通过再评审修改物质规格,人为的设置贸易技术壁垒,成为规则的制定者而非仅仅是游戏参与者。
我们都知道欧盟农药的管理体系是十分严苛的,对产品和生产商的要求都非常高。因此,如果企业的原药能通过欧盟再评审获得官方的认可,成为欧盟市场上的参考来源,这也是对产品质量的高度认可,是企业进行品牌宣传一个优质加分项。
基于以上原因,虽然原药再评审相对来说,费用比较高,周期也比较长,但对于国内农药龙头生产企业来说,这个投入是必要的。
一、欧盟原药再评审
1. 再评审简介
按照Regulation (EU) 1107/2009,欧盟农药实行周期性再评审制度。近几年再评审法规变更频繁,从最初的Regulation (EU) No 844/2012,至今经过3次重大更新,于2020年11月被Regulation (EU) 2020/1740代替。新法规主要针对活性物质批准有效期截止期在2024年3月27日以后的活性物质(不包括因截止期延期而在2024年3月27日以后过期的物质)。2024年3月27前到期的物质仍旧适用于Regulation (EU) No 844/2012。

来源:EC网站
根据再评审法规,欧盟农药活性物质的批准有效期一般为10年(不包括低风险农药和候选替代物质),在有效期截止之前便要进入活性物质再评审流程,以期在有效期到期前完成评审,实现有效期顺利更新。目前,农药企业如果计划支持再评审申请,需提交能满足Regulation (EU) 283/2013和Regulation (EU) 284/2013数据要求的全套数据,在活性物质有效期满至少3年前向欧盟提出申请,此数据要求和目前新物质批准的数据要求是一样的。
既然再评审和首次批准的数据要求是一样的,为什么申请人会需要提交新的数据呢?以下将以丙硫菌唑为例来详细说明。
根据拜耳2015年递交的再评审申请资料,丙硫菌唑的再评审卷宗在理化、分析方法、毒理、生态毒理、环境归趋、残留等方面都递交了新的资料,提交新数据的原因主要如下:
1)法规变更。丙硫菌唑在2008年按照现欧盟农药法规的前身Directive 91/414/EEC进行评审,而再评审需要按照Regulation (EC) No 1107/2009进行,因为法规的变更,数据要求也产生了变化,再评审支持者由此需提交新的数据满足新的要求。
2)指南文件变更。如分析方法部分,参考的指南文件SANCO/3030/99, SANCO/3029/99和SANCO/825/00等,在近几年欧盟都进行了更新,申请人需要按照新的指南要求来开展测试并提供相应的数据。
3)科学技术的进步,之前没法提供的数据,目前有了新的测试方法,可以完成测试报告。
4)典型制剂变化。此次再评审,除了和原物质批准相同的典型制剂,拜耳又新增了一个典型制剂,两个制剂在配方和用途上有所差异,由此需要补充新的数据。
2. 再评审新规
再评审法规的变更,也伴随着一系列执行要求的变更,基于瑞欧对欧盟法规多年的研究经验,笔者认为,目前再评审法规对申请人是更加友好的,主要体现在以下几个方面。
1)新数据通报。根据EFSA在今年3月份发布的最新指南文件, 2021年3月27以后,建议申请人为支持再评审将要开展的测试,在提交申请前至少5个月向 EFSA进行通报,不经过通报直接提交的测试报告用于支持再评审,后期有被拒绝的风险。虽然流程上变得更加繁琐了,但此举也避免了申请人开展没必要的测试或测试不符合要求的风险。
2)卷宗格式要求变化。欧盟农药登记原本的卷宗格式为CADDY,制作难度非常大,除了登记方面的专家,还需要IT介入,或者找专门的软件公司帮忙。新法规下,卷宗格式改为IUCLID,跟欧盟其他法规如REACH、BPR等采用了一样的卷宗格式,制作难度大大降低。
3)信息透明。根据欧盟《通用食品法》中关于提高评审透明度的要求,再评审的整个过程也会更加公开透明,申请人交的卷宗资料、官方的评审意见等,都会在OpenEFSA及时公开,利益相关者相比过去更加容易获取这些信息。另外,EFSA还专门开通了ConnectEFSA,以加强申请人和官方在评审过程中的良好沟通,促进再评审工作的顺利进行。
二、欧盟再评审策略分析
参与欧盟原药再评审,对各家农药企业来说,都是一个大项目,在项目启动前需要进行周密的计划。再评审需要注意哪些事项呢?笔者认为,欧盟的登记,讲究一个天时地利人和,接下来将据此进行详细分析。
1. 天时
在欧盟,新活性物质批准以后,其他原药生产者大部分时候只能通过TE评估进入市场。对于农药活性物质的再评审,欧盟有专门的工作计划,如果农药企业想要参与再评审,则必须配合官方的再评审计划表,按照计划加入。目前,再评审项目(Active Ingredient Renewal, AIR-Programme)已经有6组,一般以活性物质的有效期截止时间作为划分,具体如下。
AIR | 批准有效期 | 物质个数 | 现状 |
AIR-1 | Directive 91/414/EEC | 7 | 已经完成 |
AIR-2 | Directive 91/414/EEC | 31 | 已经完成 |
AIR-3 | 2013.1.1-2018.12.31 | 150 | 已经启动 |
AIR-4 | 2019.1.1-2021.12.31 | 215 | 已经启动 |
AIR-5 | 2022.1.1-2024.12.31 | 66 | 已经启动 |
AIR-6 | 2025.3.31-2028.12.27 | 28 | 还未启动 |
以丙硫菌唑为例,拜耳于2002年3月25日向英国提出丙硫菌唑的物质批准申请,于2008年8月1日批准通过,批准有效期为10年,截止期为2018年7月31日。根据物质有效期到期时间,丙硫菌唑属于AIR-3计划中的物质,
根据物质再评审法规,再评审申请人需在物质有效期到期前3年提出再评审申请,也就是2015年7月31日前。根据官方公布的工作文件,拜耳在2015年7月28日提交了再评审申请,再评审支持者仅拜耳一家。
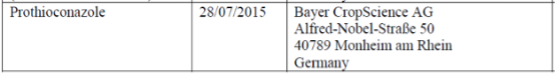
来源:EC工作文件
如果错过了再评审提交的截止期,官方就不允许申请人申请了,此时若还是希望参与再评审,只能与首家协商参与组成task force,这时决定权就在首家手里。因此,有意愿参与再评审的企业,一定要关注物质的批准有效期,及官方的AIR计划,及时向官方提交再评审申请。
按照法规,原药再评审的评审阶段需要将近三年的时间,但近几年再评审大多延期。比如丙硫菌唑再评审,虽然2015年拜耳就已经提交申请,但评审仍旧未完成,原有效期截止期延期至2022年7月31日。从EFSA评审记录来看,其在2018年10月22日到2020年3月15日暂停评审,交由企业准备进一步资料;目前,因内分泌干扰评估需要更多的数据,评审于2020年10月16日再次暂停,将于2023年4月15日之后再次启动。所以,丙硫菌唑的有效期应该还会继续延期。
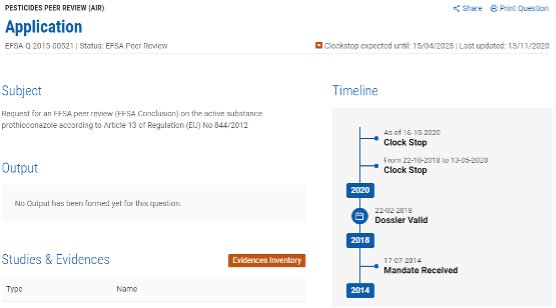
来源:EFSA网站
待原药再评审完成以后,紧跟着就是制剂再评审。还是以丙硫菌唑为例,丙硫菌唑制剂登记数量和持证人数量还是相对较多的,登记最多的国家是英国和法国,都有超过100个制剂登记,另外德国、比利时、意大利、波兰、瑞典等也均有几十个登记证,持证人除拜耳外,还有Clayton Plant Protection (UK) Limited、SAGA SAS、GRITCHE、Life Scientific Limited等几十家企业。因原药再评审延期,制剂登记有效期也会相应延期,比如丙硫菌唑,目前制剂登记在各个成员国的有效期基本都已经延期到2022年之后甚至更晚。
2. 地利
原药再评审虽然是欧盟层面执行的,但是会先有一个评估成员国(RMS)来完成卷宗的第一步评审,出具RAR(Renewal Assessment Report),再交由EFSA进行评议并出具最后的风评报告。
RMS的选择对整个评估的影响特别大。目前受到英国脱欧的影响,各成员国的工作负荷都非常大,这也是近几年欧盟再评审多有延期的一个重要原因。另外,欧盟各个成员国在评审能力方面相差巨大,并不是每一个国家都有能力进行独立评估。比如捷克,不同数据比如产品化学、毒理等的评估由不同部门进行,申请人需要与多个PM联系,由此会影响到申请人在评审过程中和RMS积极有效的沟通。不同成员国的评审风格也有所不同,如比利时对资料的要求相对来说会更加严格。最后,各成员国对评估的收费标准也不同且差异较大。同样的原药再评审,瑞典会收取最高约380万人民币的评审费,而德国在50万到130万之间。
根据法规,再评审的RMS是由欧盟指定的,申请人无法自由选择,这时候,对于RMS风格的了解就至关重要。熟悉RMS评审的“脾性”,在再评审的沟通和补资料过程中,会相对顺畅很多。
3.人和
对物质再评审来说,所谓的人和,也就是物和,就是物质不会被禁用,不至于企业费心费力好几年,一朝回到解放前。对农药登记,能否通过风险评估,一直是活性物质能否批准的一个决定因素。但欧盟不是一个完全以风险为导向的地区,在特定情况下,物质的危害对于物质能否通过批准或再评审起了决定性作用。风险导向结合危害导向,以至于欧盟近几年禁止了诸多的农药活性成分,也使欧盟成为农药登记难度极大的地区。
根据Regulation (EC) No 1107/2009,活性物质能否批准有一个cut off criteria,如果满足此标准,欧盟将不再对此活性物质进行风险评估而将直接禁止使用。具体标准如下:
1)人类健康方面,如果某物质是CMR物质(按照CLP分类,致畸致癌致突变某一项分类为1A或1B),或是内分泌干扰物,则禁用;
2)环境方面,如果是PBT, POP, 或vPvB物质,则禁用。
比如丙硫菌唑,目前就因为其代谢产物可能有潜在的内分泌干扰作用,申请人还在根据官方要求补充更多的数据,以排除其内分泌干扰危害。





 化学品合规
化学品合规
 化妆品合规
化妆品合规
 检验检测
检验检测
 安全管理智能化
安全管理智能化
 绿色低碳可持续
绿色低碳可持续
 药品合规
药品合规
 食品合规
食品合规
 食品接触材料/再生塑料
食品接触材料/再生塑料
 中国农药登记
中国农药登记
 境外农药登记
境外农药登记