

近年来,越来越多的农药企业开始关注欧盟市场,进行农药产品的布局和登记。根据欧盟农药法规,欧盟制剂产品在使用已经通过评审的原药参考来源外,还可以添加原药新来源,即使用不同厂家生产的原药。只要新来源能满足欧盟原药等同性认定的要求,也就是我们常说的欧盟原药TE(Technical Equivalence)评估,即可进入欧盟市场。由此,原药TE评估是助力企业进军欧盟市场的快捷通道!
开展欧盟原药等同性评估对企业来说,有什么好处呢?

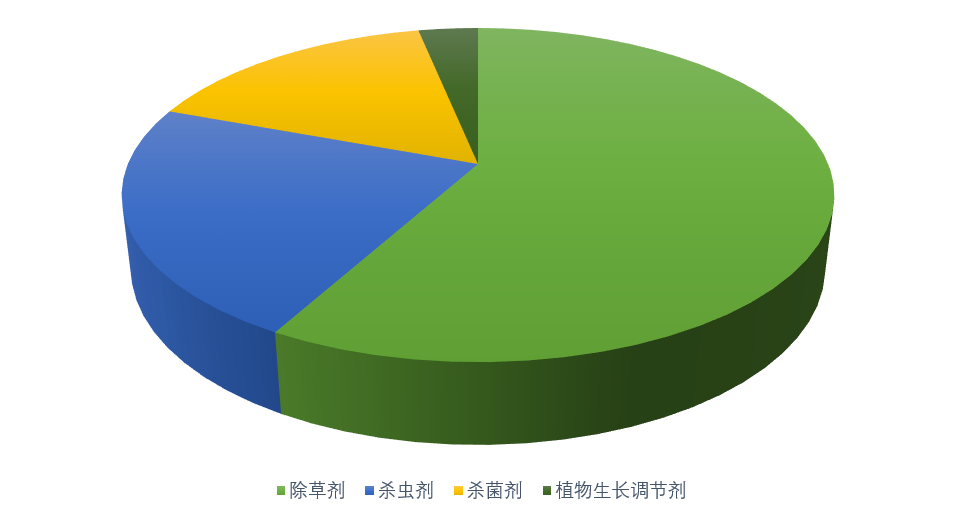
目前国内企业开展TE登记较多的产品类型产品涵盖杀虫剂、杀菌剂、除草剂、植物生长调节剂等(见下图),除草剂产品最多,杀虫剂和杀菌剂平分秋色。其中除草剂当中的热点产品有精喹禾灵、烟嘧磺隆、灭草松、炔苯酰草胺等,杀虫剂产品有吡丙醚、甲维盐、茚虫威等,杀菌剂产品有丙硫菌唑、嘧菌酯、吡唑醚菌酯、啶酰菌胺等。
结合我们多年的合规经验,开展欧盟TE评估需要注意哪些方面呢?
1. 评估成员国的选择
瑞欧农化团队深耕欧盟农药法规多年,与欧盟各个成员国保持良好的沟通合作关系。目前已经与多个成员国如英国、荷兰、比利时、法国、德国、奥地利、西班牙、瑞典等有项目合作。
根据我们的了解,英国是接收TE申请最多的成员国,其次是法国、荷兰、德国和比利时等。这些相对来说也是评审能力强、性价比高、周期快的国家,是欧盟TE评估成员国的首选。
随着英国脱欧的临近,其他成员国承接了英国较多的评估工作,评估负荷有所增加,向部分热门成员国比如比利时申请TE,目前已经需要提前预约。瑞欧建议有计划将产品投放欧盟市场的企业尽早做好准备,以免耽误产品上市时机。
2. 卷宗的准备
欧盟的TE评估分阶段进行,第一阶段主要是产品化学评估;第二阶段主要是毒理和生态毒理评估。在提交第一阶段申请资料时,应注意以下几个方面:
- 五批次样品生产日期距今最好不超过5年;
- 五批次报告要包括详细的分析方法验证部分;
- 需要关注生产工艺中可能发生的副反应,对潜在杂质形成原因做好解释;
- 若使用了高毒溶剂,其残留需要高度关注;
- 报告谱图清晰;
- 生产工艺和五批次报告信息保持一致等
如果第一阶段能够证明新来源和参考来源等同,TE评估就会通过。若新来源中出现新杂质或者现有杂质含量增加超过欧盟限量,那么TE评估就会进入第二阶段。
3. Tier II评估的应对
对于TE评估第二阶段的资料要求,欧盟官方一般要求提交QSAR报告和Ames测试报告。欧盟对这两份报告有其特定的要求,如Ames报告所用的测试样品的选择,QSAR报告评估模型及节点的选择等。瑞欧的QSAR专家在此方面有丰富的经验,目前已经针对TE项目为企业出具过几十份QSAR报告,均受到了官方的认可。
补充阅读:
什么是原药等同性评估?
原药等同性评估,通过证明新来源与欧盟已经批准的参考来源相比,危害相同或更小(活性物质含量不低于参考来源,杂质危害没有增加或增加水平在可接受范围内),这种情况下,我们认为新来源与参考来源等同。
在开展新来源与参考来源的等同性评估时,通常分为2个阶段,Tier I 和Tier II。Tier I 主要评估物质的化学组成,包括生产工艺、五批次分析、技术规格等。如果Tier I的评估不能判断新来源与参考来源是否等同,则需要进入Tier II阶段,Tier II的评估比第一阶段复杂一些,主要考量物质的毒理和生态毒理学的性质和危害。
TE评估通过之后,官方会出具一封正式批准信作为TE通过的凭证,同时会将评估报告公布在欧盟内部共享平台。
欧盟原药等同性评估是非专利原药生产企业或贸易企业,进军欧盟市场最快捷高效的方式。同时,也为后续开展制剂产品登记打下了基础。
瑞欧在欧盟农药合规领域,有丰富的经验,可以为企业定制服务,提供解决方案。企业若有进一步的疑问,欢迎咨询!
联系电话:0571-87006630
邮箱:customer@reach24h.com




 化学品合规
化学品合规
 化妆品合规
化妆品合规
 检验检测
检验检测
 安全管理智能化
安全管理智能化
 绿色低碳可持续
绿色低碳可持续
 药品合规
药品合规
 食品合规
食品合规
 食品接触材料/再生塑料
食品接触材料/再生塑料
 中国农药登记
中国农药登记
 境外农药登记
境外农药登记