

法规概述
美国药包材监管模式
FDA推出了药品主控文件(Drug Master File,DMF)的管理模式。按照不同的产品备案分类。针对不同类型DMF有不同的资料要求,主要分为以下类别:
第II类-原料药(原料药中间体及其制备所用材料或药物产品)
第III类-药包材
第IV类-药用辅料
第V类-FDA接收的参考信息
在美国,进口及内销药包材可以申请III类DMF备案。按照通用技术文件格式要求撰写资料,整理成完整的DMF文件后以电子递交的方式递交FDA备案。
美国法规中并没有强制要求药包材企业进行DMF备案,FDA对此保持中立态度。药包材的制造商可选择将技术资料直接提供给药品申请人,在药品申请资料内包含药包材信息;或者选择DMF备案直接向FDA提交资料,确保相关的机密信息不向下游泄露。
监管部门及其职责
美国食品药品监督局(FDA)是直属于美国卫生公共服务部的联邦政府机构,是美国进行药品安全监管主体。FDA的监管主要针对药品,而对于药品相关的“附属”产品(如原料药、药用辅料和药包材)可以通过药品主控文件(Drug Master File,DMF)的管理模式实行关联审评。
药品评价和研究中心(CDER)隶属于FDA,目前是FDA最大的一个审评中心,该机构主要负责:监管处方和非处方药, 对新药、仿制药进行上市前评估, 并负责药物的安全性、质量以及有效性及进行关联评审的相关工作。
法规事务办公室(Office of Regulatory Affairs,ORA)隶属于FDA,主要负责检查及调查受管制的产品和制造商,对受管制的产品进行抽样分析,同时对进口至美国产品检查、突发事件处理、召回及强制执行等。
备案流程
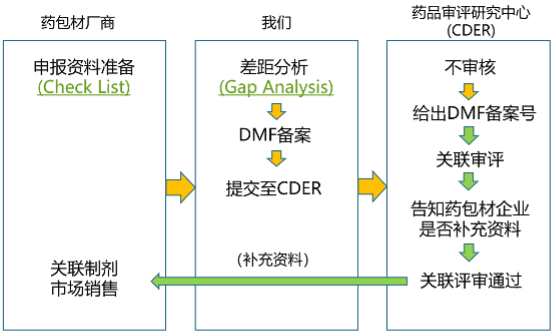
药包材DMF备案步骤
药包材企业在FDA网站提交DMF电子版资料;
药品评审研究中心(CDER)不对登记资料审查,在2~3周内给出DMF备案号;
CDER结合药品进行关联审评,若药包材相关资料不完整时,会告知药包材企业进行补充直至完整(对药包材企业不存在批准或不批准);
关联审评通过后告知药包材企业。
药包材备案资料
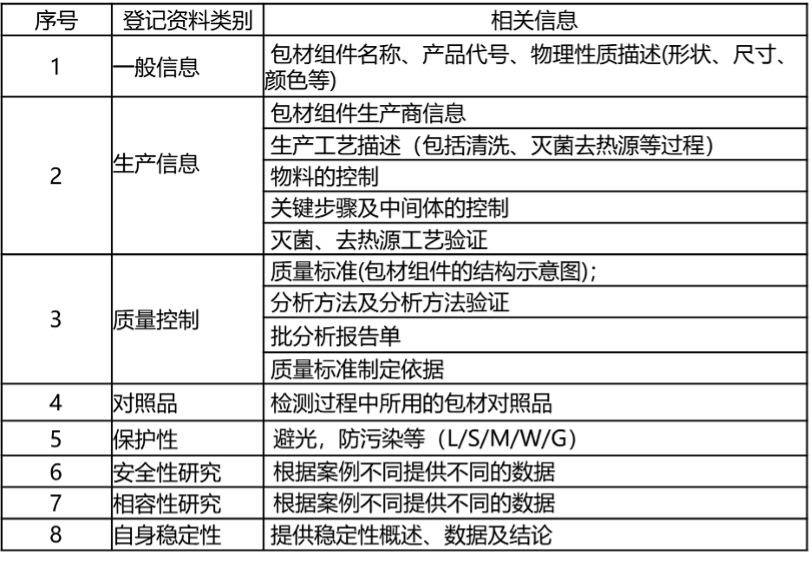
关于药包材的药品主控文件(Drug Master File,DMF),是呈交FDA的存档待审资料,资料内容包括药包材的一般信息、自身性质以及生产信息和质量控制信息等内容:
我们的服务
药包材DMF备案咨询
药包材DMF备案业务代理
备案资料翻译
药包材关联评审跟进
药包材DMF年度报告撰写
药包材DMF 授权书(LOA)撰写
终止或重启DMF申报
实验委托及测试监理
官方问询与沟通
我们的优势
超过13年法规研究和包材注册申报工作经验;
曾经参与申报的包材包括:玻璃输液瓶、塑料输液瓶、粉液双室袋、液液多室袋、组合盖、胶塞、异戊二烯垫片、预灌封注射器、注射剂西林瓶、口服液体塑料瓶、口服固体塑料瓶、口服固体复合膜、塑料安瓿、玻璃安瓿等等。



 化学品
化学品
 食品接触材料
食品接触材料
 化妆品
化妆品
 绿色双碳
绿色双碳
 境外农药登记
境外农药登记
 中国农药登记
中国农药登记