Generic Drug Application (ANDA) in China
Generic drugs comprise a significant part of China\’s pharmaceutical market. In 2020, 63% of marketed drugs in China were generics. The Chinese generic drug market size reached 970.7 billion yuan (143.85 billion USD) in 2019 and fell to 808.7 billion yuan1 (circa 119.84 billion USD) in 2020 due to the economic slowdown brought by the COVID-19 pandemic.
However, China\’s generic drug market is expected to grow. One reason is that generics usually have lower prices than patented originator drugs, so generics have advantages in volume-based procurement where the price competition is fierce.
Another reason is that generics\’ quality has improved remarkably since 2015 as China mandates that generics should be equivalent to originator drug/reference listed drug in quality and therapeutic effects.
Furthermore, China encourages the development of clinically-needed and high-quality generics. In the drug patent linkage system, China awards the first generic to successfully challenge the relevant patent drug with 12-month market exclusivity. Thus, generics have remarkable market potential in China.
This article introduces China\’s regulations on generic drug marketing authorization application, also known as abbreviated new drug application (ANDA).
Contents
- Definition and Classification of Generic Drugs
- Reference Listed Drug (RLD)
- Quality and Therapeutic Equivalence
- ANDA Dossier
- ANDA Procedures
- ANDA Fees
- BaiPharm Service of Generic Drug Registration
1. Definition and Classification of Generic Drugs
Generic drugs are copies that are equivalent to originator drugs in dose, safety, strength, quality, efficacy, and indication. In China, there are five general classes of chemical drugs. Generic chemical drugs are divided into Class 3, Class 4, and Class 5.2. Generic drugs only refer to chemical drug copycats, not including biosimilars.
| Classification of Generic Drugs2 | |
| Class 3 domestic generic drug manufactured in China | The relevant originator drug/RLD has been approved overseas only, but has not been approved in China;
The generic drug has the same active pharmaceutical ingredient (API), dosage form, strength, indications, administration route, and dosage with the RLD. The generic drug can demonstrate quality and therapeutic equivalence to the RLD. * The generic drug\’s strength and dosage can be different from those of RLD if sufficient and reasonable data support the differences. |
| Class 4 domestic generic drug manufactured in China | The relevant originator drug/RLD has been approved in China;
The generic drug has the same API, dosage form, strength, indications, administration route, and dosage with the RLD. The generic drug can demonstrate quality and therapeutic equivalence to the RLD. |
| Class 5.2 overseas generic drug imported to China | The generic drug has been approved overseas;
The generic drug has the same API, dosage form, strength, indications, administration route, and dosage with the RLD. The generic drug can demonstrate quality and therapeutic equivalence to the RLD. |
Notes:
* Original drug refers to the first drug that obtains Chinese or overseas marketing authorization supported by sufficient safety and efficacy data.
* For a generic drug in the joint research in China and overseas but manufactured overseas, it should be registered as a Class 5.2 generic drug. If such a drug applies for clinical trials in China, it is not required to provide the overseas certificate of pharmaceutical product (CPP).
2. Reference Listed Drug (RLD)
A generic drug applicant should choose an appropriate RLD to approve the generic drug\’s equivalence to the RLD in quality and therapeutic efficacy.
(1) NMPA recommends drug applicants to choose RLDs in the following order of priority: originator drug approved in China → drug manufactured by the confirmed foreign originator pharmaceutical company in China or manufactured with technology transferred from overseas to China → originator drug not imported to China.
(2) When the production of the originator drug is discontinued, or the originator drug is not appropriate to be an RLD, the generic drug applicant can choose an internationally recognized drug as the RLD. Internationally recognized drug refers to the drug that has received marketing approval and RLD status in countries and regions with standardized management, such as the U.S., Japan, and the EU.
(3) Other drugs can be chosen if they are confirmed by the NMPA to be qualified in safety, efficacy, and quality controllability.
China NMPA has released the Catalog of Reference Listed Drugs and will continue selecting and adding drugs to the catalog. To add a new RLD to the catalog, a generic drug applicant, drug research/manufacturing company, or industry association should submit an RLD application to the Center for Drug Evaluation (CDE), an office under NMPA.
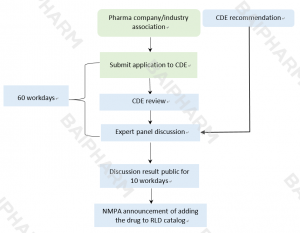
RLD Application Procedures in China
3. Quality and Therapeutic Equivalence
Generic drugs need to be equivalent to the originator drug/RLD in both in vivo and in vitro performances:
3.1 Pharmaceutical equivalence for in Vitro performance:
Generic drugs must have the same API, dosage, administration route, and dosage form with the originator drug/RLD, and comply with drug quality standards on API content, purity, homogeneity, disintegration time, dissolution profile, etc.
In terms of non-API ingredients, generics do not have to be the same as the originators/RLDs. But for special cases, for example, an injection\’s excipient needs to have the same type and amount as those of the originator/RLD.
3.2 Therapeutic equivalence for in vivo performance:
For oral generic drugs, bioequivalence (BE) studies usually suffice to prove their therapeutic equivalence. Detailed BE requirements are specified in CDE\’s guidelines, e.g., Bioequivalence Guidelines on Ebastine Tablets.
However, for special oral drugs and complex dosage forms, clinical trials are required to provide efficacy data.
In 2018, NMPA made No. 32 and No. 136 Announcements to allow trial simplification or exemption of BE studies for some drugs, e.g., BCS (Biopharmaceutics Classification System) I or III oral drugs. Applicants of the listed drugs should conduct comparative chemistry, manufacturing, and controls (CMC) study and other required research according to the Guidelines on Exempting BE Studies for Human Body. If the drug is qualified, the applicants can submit BE study exemption application with the ANDA.
Contact Us
BaiPharm, a subsidiary of REACH24H Consulting Group, is specialized in pharmaceutical industry consultancy services. With our experts across China, Japan, South Korea, the United States, and Europe, BaiPharm is fully qualified to engage in complex market entry projects of finished drugs, APIs, excipients, and packaging materials for most key markets.
If you have any questions about the market approval of generic drugs, please feel free to contact us.
Tel: +86 (0)571-87007555
Email: customer@reach24h.com
